基本信息
药品名称
商品名:开浦兰
汉语拼音:Zuoyilaxitan Pian
通用名称:左乙拉西坦片
英文名:Levetiracetam Tablets
成份
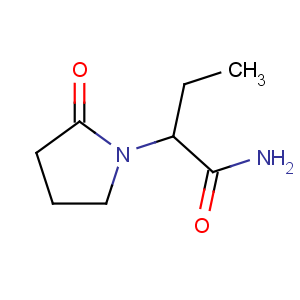
左乙拉西坦的化学名称为(S)-α-乙基-2-氧代-1-吡咯烷乙酰胺,分子式 :C8H14N2O2,分子量 :170.21。

[1]
用法用量
给药途径
口服。需以适量的水吞服,服用不受进食影响。
给药方法和剂量
成人(>18岁)和青少年(12-17岁)(体重≥ 50 kg者) :起始治疗剂量为每次500 mg,每日2次。根据临床效果及耐受性,每日剂量可增加至每次1500 mg,每日2次。剂量的变化应每2-4周增加或减少500 mg/次,每日2次。
老年人 (≥ 65岁):根据肾功能状况,调整剂量 (详见下文有关肾功能受损病人描述)。
4-11岁的儿童和青少年(12-17岁)(体重≤ 50 kg者) :起始治疗剂量是10 mg/kg,每日2次。根据临床效果及耐受性,剂量可以增加至30 mg/kg,每日2次。 剂量变化应以每2周增加或减少10 mg/kg,每日2次。应尽量使用最低有效剂量。儿童和青少年体重≥ 50 kg者,剂量和成人一致。青少年和儿童推荐剂量:起始剂量10 mg/kg,每日2次,最大剂量30 mg/kg,每日2次。体重15 kg:起始剂量每次150 mg,每日2次,最大剂量每次450 mg,每日2次。体重20 kg:起始剂量每次200 mg,每日2次,最大剂量每次600 mg,每日2次。体重25 kg:起始剂量每次250 mg,每日2次,最大剂量每次750 mg,每日2次。体重50 kg或以上:起始剂量每次500 mg,每日2次,最大剂量每次1500 mg,每日2次。20 kg 以下的儿童,为精确调整剂量,起始治疗应使用口服溶液。
婴儿和小于4岁的儿童患者:目前尚无相关的充足的资料。
肾功能受损的病人 :成人肾功能受损病人,根据肾功能状况,按表中不同肌酐清除率(CLcr) mL/min(测出血清肌酐值按下述计算方法)调整日剂量。CLcr=140-年龄(岁) x 体重(kg)/72 x 血清肌酐值(mg/dl)女性病人:上述计算值 x 0.85<BR>肾功能受损病人的剂量正常病人(肌酐清除率80 mL/min) :每次500-1500 mg,每日2次。轻度异常(肌酐清除率50-79 mL/min) :每次500-1000 mg,每日2次。中度异常(肌酐清除率30-49 mL/min) :每次250-750 mg,每日2次。严重异常(肌酐清除率<30 mL/min) :每次250-500 mg,每日2次。正在进行透析晚期肾病病人 :500-1000 mg ,每日1次。服用第1天推荐负荷剂量为左乙拉西坦750 mg。透析后,推荐给予250-500 mg附加剂量。儿童肾功能损害病人应根据肾功能状态调整剂量,因为左乙拉西坦的清除与肾功能有关。
肝病患者:对于轻度和中度肝功能受损的病人,无需调整给药剂量。严重肝损的病人,根据肌酐清除率可能低估肾功能不全的程度,因此,如果病人的肌酐清除率小于70 mL/min,日剂量应减半。
不良反应
成人临床
研究汇总的安全性数据表明, 药物组和安慰剂组不良反应的发生率相似,分别为46.4%和42.2%。 其中,严重不良反应分别为2.4%和2.0%。最常见的不良反应有嗜睡,乏力和头晕,常发生在治疗的开始阶段。随时间的推移,中枢神经系统相关的不良反应发生率和严重程度会随之降低。左乙拉西坦不良反应没有明显的剂量相关性。
儿童临床
研究(4-16岁)表明药物组和安慰剂组产生不良反应的发生率相似,分别为55.4%和40.2%,药物组未发生严重不良反应(安慰剂组1.0%)。儿童最常见的不良反应有嗜睡、敌意、神经质、情绪不稳、易激动、食欲减退、乏力和头痛。除行为和精神方面不良反应发生率较成人高(儿童38.6%,成人 18.6%)外,总的安全性和成人相仿。成人和儿童不良反应的风险是具有可比性的。总结成人和儿童临床研究结果和上市后经验,评估了每个系统的不良反应和发生频率 :很常见>10% ;常见1-10% ;少见0.1-1% ;罕见:0.01-0.1% ;非常罕见<0.01%,包括单独的报告。上市后临床应用的数据,尚不足以估计治疗人群中不良反应的发生率。
症状汇总
- 全身反应和给药部位不适 :很常见乏力。
- 神经系统不适 :很常见嗜睡,常见健忘、共济失调、惊厥、头晕、头痛、运动过度、震颤。
- 精神心理变化 :常见易激动、抑郁、情绪不稳、敌意、失眠、神经质、人格改变、思维异常。上市后不良事件报道 :行为异常、攻击性、易怒、焦虑、错乱、幻觉、易激动、精神异常、自杀、自杀性意念、自杀企图。但还没有足够数据,用于估计对它们的发生率或建立因果关系。
-消化道不适 :常见腹泻、消化不良、恶心、呕吐。
- 代谢和营养障碍 :常见食欲减退。当病人同时服用托吡酯时,食欲减退的危险性增加。
- 耳及迷路系统不适 :常见眩晕。
- 眼部不适 :常见复视。
- 伤害、中毒和后续的并发症 :常见意外伤害。
- 感染和传染 :常见感染。
- 呼吸系统不适 :常见咳嗽增加。
- 皮肤和皮下组织异常变化 :常见皮疹。上市后不良事件报道 - 脱发,某些病例中停药后自行恢复。
- 血液系统和淋巴系统异常变化 :上市不良事件报道 - 白细胞减少、嗜中性细胞减少、全血细胞减少、血小板减少,但还没有足够数据,用于估计它们发生率或建立因果关系。
禁忌症
对左乙拉西坦过敏或者对吡咯烷酮衍生物或者其他任何成分过敏的病人禁用。
注意事项
根据当前的临床实践,如需停止服用本品,建议逐渐停药。(例如 :成人每隔2-4周,每次减少500 mg,每日2次 ;儿童应每隔2周,每次减少10 mg/kg,每日2次)。临床研究中,一些患者对加用左乙拉西坦治疗有效应,可以停止原合并应用的抗癫痫药物(研究中共有69位患者其中的36位成人患者)。
临床研究中报告有14%服用左乙拉西坦的成人及儿童患者癫痫发作频率增加25%以上,但在服用安慰剂的成人及儿童患者中,也各有26%及21%患者癫痫发作频率增加。对于肝功能损害的病人,参照[用法与用量]。对于严重肝功能损害的病人,应先行检查肾功能,然后进行调整。
对驾驶和应用机器影响 :目前没有研究关于服药后对机器驾驭能力和驾驶车辆能力的影响。
由于个体敏感性差异,在治疗初始阶段或者剂量增加后,会产生嗜睡或者其他中枢神经症状。因而,对于这些需要服用药物的病人,不推荐操作需要技巧的机器,如驾驶汽车或者操纵机械。
孕妇及哺乳期妇女用药
目前没有孕妇服用本品的资料,动物试验证明该药有一定的生殖毒性。对于人类潜在的危险目前尚不明确。如非必要,孕妇请勿应用左乙拉西坦。 突然中断抗癫痫治疗,可能使病情恶化, 对母亲和胎儿同样有害。动物试验表明左乙拉西坦可以从乳汁中排出,所以,不建议病人在服药同时哺乳。
儿童用药
见[用法用量]项。
老年患者用药
见[用法用量]项。
药物相互作用
体外数据显示:治疗剂量范围内获得的高于Cmax水平的浓度时左乙拉西坦及其主要代谢物,既不是人体肝脏细胞色素P450、环氧化水解酶或尿苷二磷酸-葡萄苷酶的抑制剂,也不是它们具有高亲合力的底物。因此,不易出现药代动力学相互作用。另外,左乙拉西坦不影响丙戊酸的体外葡萄苷酶作用。
左乙拉西坦血浆蛋白结合率低(<10%),不易产生因与其他药物竞争蛋白结合位点所致临床显著性的相互作用。
临床药代动力学研究(苯妥英、丙戊酸钠、口服避孕药、地高辛、华法令和丙磺舒)和安慰剂对照临床试验中,通过药代动力学筛选评估了药物之间的潜在药代动力学相互作用。
左乙拉西坦和其他抗癫痫药物(AEDs)间的药物-药物相互作用 :苯妥英+左乙拉西坦(每日3000 mg)对难治性的癫痫病人,苯妥英药代动力学特性不产生作用。苯妥英的应用也不影响本品的药代动力学特性。
丙戊酸钠+左乙拉西坦(1500 mg,每日2次)不改变健康志愿者丙戊酸钠药代动力学特性。丙戊酸钠500 mg,每日2次,不改变左乙拉西坦吸收的速率或程度,或其血浆清除率,或尿液排泄。也不影响主要代谢物的暴露水平和排泄。
对安慰剂对照临床研究获得的左乙拉西坦和其他抗癫痫药物(卡马西平、加巴喷丁、拉莫三嗪、苯巴比妥、苯妥英、去氧苯巴比妥和丙戊酸钠)的血清浓度进行了评估,数据显示左乙拉西坦不影响其他抗癫痫药物的血药浓度。这些常用的抗癫痫药物也不影响本品药代动力学特性。
儿童病人抗癫痫药物的作用同时服用酶诱导型抗癫痫药,本品体内表观总清除率增加约22%。但无需进行剂量调整。左乙拉西坦不影响卡马西平、丙戊酸钠、托吡酯或拉莫三嗪的血浆药物浓度。
其他药物相互作用 :口服避孕药+服用左乙拉西坦(500 mg,每日2次)不影响含有0.03 mg炔雌醇和0.15 mg左炔诺孕酮口服避孕药的药代动力学特性,或促黄体激素和黄体酮含量水平,表明本品不影响避孕药功效。应用口服避孕药并不影响本品的药代学特性。
地高辛+服用左乙拉西坦(1000 mg,每日2次)不影响每日剂量0.25 mg地高辛的药代动力学和药效学(ECG)特性。应用地高辛并不影响本品的药代学特性。
华法令+服用左乙拉西坦(1000 mg,每日2次)不影响R和S型华法令的药代动力学特性。凝血时间不受左乙拉西坦影响。应用华法令并不影响本品的药代学特性。
丙磺舒(500 mg,每日4次)为肾小管分泌阻滞剂,会抑制左乙拉西坦的主要代谢物的肾脏清除率,但不是左乙拉西坦药代学特性(1000 mg,每日2次),但这些代谢物的浓度很低。其他需经肾小管分泌清除的药物也会影响代谢物的肾脏清除。目前无左乙拉西坦合并丙磺舒用药的研究,左乙拉西坦合并应用其他主动分泌药物对药效影响(例如非甾体抗炎药、磺胺药和甲氨蝶呤),尚不明确。
药物过量
症状
据观察有嗜睡、激动、攻击性、意识水平下降、呼吸抑制及昏迷。
急救措施
在急性药物过量后,应采取催吐或洗胃使胃排空。目前尚无左乙拉西坦的解毒剂。治疗需对症治疗,也可包括血液透析。透析排出的效果 :左乙拉西坦60%,主代谢产物74%。
临床试验
临床研究
对难治性癫痫部分性发作病人(伴或不伴有继发性全身发作)进行的3个多中心、随机、双盲和安慰剂对照的临床研究,建立了成年人左乙拉西坦作为加用治疗(其他抗癫痫药物的辅助治疗)的有效性数据。进入研究1或研究2的病人患有至少2年的难治性癫痫部分性发作,并服用2种以上传统抗癫痫药。进入研究3的病人患有至少1年的难治性癫痫部分性发作,并服用1种传统抗癫痫药。在试验时,病人正服用至少1个最多2个抗癫痫药的稳定的给药方案。在基线阶段,每个4周的阶段病人必须至少发生2次部分性发作。
临床研究1 :是在美国41个研究地点进行的双盲、安慰剂对照的平行试验,在一个12周基线阶段后,病人随机分配入左乙拉西坦剂量1000 mg/天(N=97)、左乙拉西坦3000 mg/天(N=101)和安慰剂(N=95)组每日分2次给药。进行18周的治疗周期内(6周的逐量加药期+12周的评价期)的疗效评估,试验期间可同服抗癫痫药治疗方案保持不变。主要的疗效指标指在整个随机治疗阶段(逐量加药阶段+评价阶段)相对于安慰剂组每周部分性发作频度下降百分比的组间比较。次要的疗效指标包括有效应答率(部分性发作频度下降≥ (greater than or equal to) 50%的病人发生率)。结果显示 :左乙拉西坦治疗组每周部分性发作频度有明显地减少。整个随机治疗阶段左乙拉西坦治疗组的有效应答率明显高于安慰剂组,并呈剂量相关性。左乙拉西坦1000 mg/天(N=97)组 :相对于安慰剂组部分性发作频度下降百分比为26.1% ;左乙拉西坦3000 mg/天(N=101)组 :相对于安慰剂组部分性发作频度下降百分比为30.1%,P<0.001。
临床研究2:是在欧洲62个研究中心进行的双盲、安慰剂对照的交叉试验,设计试验的第1阶段(阶段A)作为平行组试验来分析,在12周的基线阶段后,病人随机分配入左乙拉西坦1000 mg/天(N=106)、左乙拉西坦2000 mg/天(N=105)和安慰剂(N=111)组,每日分2次给药。进行了16周的治疗周期(含4周的逐量加药期和12周的稳定剂量评价阶段)的疗效评估,在试验期间同服抗癫痫药的治疗方案保持不变。主要的疗效指标在整个随机治疗阶段(逐量加药期+评价阶段)相对于安慰剂组每周部分性发作频度下降百分比的组间比较,次要的疗效指标包括有效应答率(部分性发作频度下降≥ (greater than or equal to) 50%的病人发生率)。结果显示 :左乙拉西坦治疗组每周部分性发作频度有明显地减少。整个随机治疗阶段左乙拉西坦治疗组的有效应答率明显高于安慰剂组,并呈剂量相关性。左乙拉西坦2000 mg/天对左乙拉西坦1000 mg/天有效应答率的比较具统计显著性(P=0.02),和交叉试验的分析产生了相似的结果。左乙拉西坦1000 mg/天(N=106)组 :相对于安慰剂组部分性发作频度下降百分比为17.1% ;左乙拉西坦2000 mg/天(N=105)组 :相对于安慰剂组部分性发作频度下降百分比为21.4%,P<0.001。
临床研究3是在欧洲47个研究中心进行的双盲、安慰剂对照的平行组试验,该研究比较了对伴有或不伴有继发性全身发作的难治性癫痫部分性发作病人,仅同服一种抗癫痫药治疗,12周的基线阶段后,病人随机分配入的上述2个治疗组中的一个。16周的治疗周期由一个4周的逐量加药阶段,在12周的稳定剂量阶段进行了疗效的评估。主要的疗效指标在整个治疗阶段(逐量加药期+评价阶段)相对于安慰剂组每周部分性发作频度下降百分比的组间比较,次要的疗效指标包括有效应答率(部分性发作频度下降≥ (greater than or equal to) 50%的病人发生率)。结果显示 :左乙拉西坦治疗组每周部分性发作频度有明显地减少。整个随机治疗阶段左乙拉西坦治疗组的有效应答率明显高于安慰剂组。左乙拉西坦3000 mg/天(N=180) :相对于安慰剂组部分性发作频度下降百分比为23.0%,P<0.001。
有效性的研究
北美60个试验中心对4-16岁难治性癫痫部分性发作的儿童进行多中心、随机、双盲和安慰剂对照的临床研究,建立了在儿童患者中左乙拉西坦作为加用治疗(其他抗癫痫药物的辅助治疗)的有效性数据。患儿在筛选前4周内至少有4次发作,在8周基线期内(每个4周的基线期内都至少有4次发作),同时服用1-2种稳定剂量的抗癫痫药。共有198名患儿随机分配入(左乙拉西坦组N=101和安慰剂组N=97)。左乙拉西坦起始剂量为20 mg/kg/天,分2次给药,在治疗期间,以20 mg/kg/天的增量进行调整,在2周的时间内可增至目标剂量60 mg/kg/天。本临床研究含8周的基线期、4周逐量加药期,以及10周的评价期。主要的疗效指标指在整个随机治疗阶段(逐量加药阶段+评价阶段)相对于安慰剂组每周部分性发作频度下降百分比的组间比较。次要的疗效指标包括有效应答率(部分性发作频度下降≥ (greater than or equal to) 50%的病人发生率)。结果显示 :左乙拉西坦治疗组每周部分性发作频度有明显地减少。整个随机治疗阶段左乙拉西坦治疗组的有效应答率明显高于安慰剂组。左乙拉西坦(N=101) :相对于安慰剂组部分性发作频度下降百分比为26.8%,P<0.0002。
中国临床研究
在中国6个中心(上海、北京、重庆、成都),对左乙拉西坦口服16周作为添加治疗成人及16岁以上青少年癫痫部分性发作的疗效与安全性进行了多中心、随机、双盲平行、安慰剂对照的临床研究,两个治疗组分别为安慰剂组和左乙拉西坦组日剂量为3000 mg。16周的临床试验共筛选了224例患者,最终189例完成了试验(左乙拉西坦组98例,安慰剂组91例)。所有受试者均为中国人,男女比例分别52%和48%。两治疗组人口统计学资料和其他基线特征具有可比性,基线期的每周癫痫发作频率也相似(左乙拉西坦组为1.81次/周,安慰剂组1.75次/周)。主要疗效指标为16周治疗期间(4周增量期+12周维持治疗期)每周癫痫部分性发作(I型)频率。16周试验结束时研究者采用整体临床疗效评估表对受试者整体疗效进行了评价。意向治疗人群分析结果表明,16周治疗期内左乙拉西坦组每周癫痫部分性发作频率较安慰剂组明显下降(p<0.001),疗效明显优于安慰剂,其相对安慰剂组减少百分数为26.8%(95%可信区间 :14.0-37.7%)。方案治疗人群结果类似。16周治疗期内,左乙拉西坦组的癫痫部分性发作50%有效率的比例为57/102(55.9%),明显高于安慰剂组的26/100(26.0%)。相对安慰剂的OR值为3.6(95% 可信区间 :2.0-6.5,p<0.001)。左乙拉西坦组中有11例(10.8%)未发生任何癫痫部分性发作,明显高于安慰剂组(2例,2.0%,p<0.001)。本研究安全性评估结果显示安慰剂组和左乙拉西坦组是有可比性的。左乙拉西坦组最常报导的不良事件是嗜睡(18个受试者,17.5%),其次是血小板降低。疗效/药代动力学/药效学研究结果 :意向治疗(ITT)人群每周癫痫部分性发作次数频率分析
(a) 将每周癫痫部分性发作的频率经自然对数转换[Ln(1+X)]后,采用协方差模型进行分析,以基线值为协变量,以研究中心和治疗组为固定变量进行估计。(b) 相对安慰剂组减少百分数采用以下公式进行计算 :100 × [1 - Exp (LSM左乙拉西坦 - LSM安慰剂)]。(c) 与安慰剂进行比较。(d) 采用WILCOXON秩和检验。(e)采用WILCOXON秩和检验。(f) 采用Logistic回归分析。(g)采用CMH以中心分层分析。总的来说,左乙拉西坦作为加药疗法治疗成年人及16岁以上青少年癫痫部分性发作,16周治疗期内可以显著减少癫痫每周发作频率,安全耐受性较好。
药理作用
左乙拉西坦是一种吡咯烷酮衍生物,其化学结构与现有的抗癫痫药物无相关性。左乙拉西坦抗癫痫作用的确切机制尚不清楚。在多种癫痫动物模型中评估了左乙拉西坦的抗癫痫作用。左乙拉西坦对电流或多种致惊剂最大刺激诱导的单纯癫痫发作无抑制作用,并在亚最大刺激和阈值试验中仅显示微弱活性。但对毛果芸香碱和红藻氨酸诱导的局灶性发作继发的全身性发作观察到保护作用,这两种化学致惊厥剂能模仿一些人伴有继发性全身发作的复杂部分性发作的特性。左乙拉西坦对复杂部分性发作的大鼠点燃模型的点燃过程和点燃状态均具有抑制作用。这些动物模型对人体特定类型癫痫的预测价值尚不明确。体外、体内试验显示,左乙拉西坦抑制海马癫痫样突发放电,而对正常神经元兴奋性无影响,提示左乙拉西坦可能选择性地抑制癫痫样突发放电的超同步性和癫痫发作的传播。左乙拉西坦在浓度高至10 uM时,对多种已知受体无亲和力,如苯二氮类、GABA、甘氨酸、NMDA、再摄取位点和第二信使系统。体外试验显示左乙拉西坦对神经元电压门控的钠离子通道或T-型钙电流无影响。左乙拉西坦并不直接易化GABA能神经传递,但研究显示对培养的神经元GABA和甘氨酸门控电流负调节子活性有对抗作用。在大鼠脑组织中发现了左乙拉西坦的可饱和的和立体选择性的神经元结合位点,但该结合位点鉴定和功能目前尚不明确。
毒理研究
遗传毒性
左乙拉西坦Ames试验、CHO/HGPRT基因座哺乳动物细胞基因突变试验、CHO细胞染色体畸变试验、小鼠微核试验结果均为阴性。左乙拉西坦水解产物和主要人体代谢物Ames试验、小鼠淋巴瘤试验结果均为阴性。
生殖毒性
在剂量高达1800 mg/kg/天[按mg/m2或暴露量(AUC)推算相当于人最高推荐剂量(MRHD)3000 mg的6倍]时,未见对雄性或雌性大鼠生育力或生殖行为的不良影响。
在动物试验中,左乙拉西坦在相似于或高于人体治疗剂量时可产生发育毒性。妊娠大鼠在器官形成期给药,剂量为3600 mg/kg/天(以mg/m2推算相当于MRHD的12倍)时,可见胎仔体重降低和胎仔骨骼变异发生率增加。发育毒性的无影响剂量为1200 mg/kg/天,该试验中未见母体毒性。妊娠家兔在器官形成期给药,剂量为≥ (greater than or equal to) 600 mg/kg/天(以mg/m2推算相当于MRHD的4倍)时,可见胚胎-胎仔死亡率增加,胎仔骨骼异常发生率增加 ;在剂量为1800 mg/kg/天(按mg/m2推算相当于MRHD的12倍)时,可见胎仔体重降低,胎仔畸形发生率增加,并可见母体毒性。发育毒性的无影响剂量为200 mg/kg/天。
雌性大鼠在妊娠和哺乳期间给药,在剂量为≥ (greater than or equal to) 350 mg/kg/天(按mg/m2推算相当于MRHD)时,可见胎仔骨骼异常、出生前和/或出生后生长迟滞 ;在剂量为1800 mg/kg/天(按mg/m2推算相当于MRHD的6倍)时,可见幼仔死亡率增加和子代行为异常 ;该试验中发育毒性的无影响剂量为70 mg/kg/天,未见明显的母体毒性。大鼠在妊娠第3期和整个哺乳期给药,剂量至1800 mg/kg/天(按mg/m2推算相当于MRHD的6倍),未见对发育和母体的不良影响。
致癌性
大鼠掺食法给予左乙拉西坦104周,剂量为50、300和1800 mg/kg/天(高剂量按mg/m2或暴露量推算相当于MRHD的6倍),未见致癌性。小鼠掺食法给予左乙拉西坦80周,剂量为60、240和960 mg/kg/天(高剂量按mg/m2或暴露量推算相当于MRHD的2倍),未见致癌性。但由于给药剂量较低,未能充分评估对该动物种属的潜在致癌性。
药代动力学
左乙拉西坦是极易于溶解和具有高度渗透性化合物。呈线性代谢,个体内和个体间差异小。多次给药不影响其清除率。本品没有性别、种族差异性和生理节奏差异。本品的药代动力学研究显示,健康志愿者和病人的药代动力学数据具有可比性。由于左乙拉西坦的吸收完全性和线性关系,其血药浓度可以根据口服剂量mg/kg进行预测,因而没有必要对左乙拉西坦的进行血药浓度监控。成人及儿童患者的唾液和血药浓度显示有显著的相关性(服用本品片剂或本品液体制剂4小时后,唾液/血液药物浓度比是1-1.7)。
成人和青少年吸收
左乙拉西坦经口服后迅速吸收,口服绝对生物利用度接近100%。给药1.3小时后血药浓度达峰,如果每日给药2次,2天后达到稳态坪浓度,如果单剂量为1000 mg及1000 mg,每日2次,典型的峰浓度为31和43 ug/mL。吸收时间与剂量无关,摄取食物不影响吸收速度。
分布
目前没有人体组织分布的数据。无论是左乙拉西坦还是其主要代谢产物均不易与血浆蛋白结合(<10%)。分布容积为0.5-0.7 L/kg,接近人体水容积。
生物转运
左乙拉西坦在人体内并不广泛分解,主要代谢途径是通过水解酶的乙酰胺化 (给药剂量的24%)。主要代谢产物UCB L057并不由肝色素P450转运体系转化而来。体内大部分组织包括血细胞均可测定乙酰胺基团水解物。代谢产物UCB L057无药理活性。2个少量代谢的途径也已经确定,一个是羟化吡咯烷途径 (给药剂量的1.6%),另外是吡咯烷基团开环,大约占剂量的0.9%。其他不能够确定的代谢途径的代谢产物占给药剂量的0.6%。目前体外试验数据表明,无论是左乙拉西坦还是其主要代谢物均无首过效应。体外试验数据表明左乙拉西坦和其主要代谢产物并不抑制肝色素P450异构(CYP3A4,2A6,2X8/9/10,2C19,2D6,2E1和1A2)葡萄糖醛转移酶(UGT1*6,UGT1*1和UGT[pI6.2]和环氧化物羟基酶活性。此外,左乙拉西坦在体外试验表明不影响丙戊酸的葡萄糖醛化。在人体肝细胞组织中,左乙拉西坦不产生酶诱导作用。因而,本品和其他物质共同应用,通常不会产生相互作用,反之亦然。
消除
成人血浆半衰期: 7±1小时,并不因给药剂量不同、给药途径不同或者重复给药而更改。平均体内总清除率为0.96 mL/min/kg。药物主要从尿液中排泄,约为剂量的95%(大约93%在48小时内排泄)。从粪便内排泄的药物仅仅占0.3%。在开始给药的48小时内,累计左乙拉西坦和其代谢产物的排泄率分别为给药剂量的66%和24%。左乙拉西坦和UCB L057肾脏清除率分别为0.6和4.2 mL/min/kg, 这表明左乙拉西坦通过肾小球滤过后经肾小管重吸收后排除,主要代谢产物也是通过肾小管分泌和肾小球滤过消除。左乙拉西坦的消除率和肌老年病人 :老年患者左乙拉西坦的半衰期大约延长了40%(10-11小时)。 这与肾脏功能下降有关。
儿童(4-12岁)单剂量给药(20 mg/kg)的左乙拉西坦血浆半衰期为6.0小时(6-12岁)。其表观清除率(体重调节后)约比癫痫成人高30%。童(4-12岁)重复口服(20-60 mg/kg/日)后,左乙拉西坦迅速吸收。用药后0.5-1小时达峰浓度。峰浓度及曲线下面积呈线性,并与剂量成比例增加。清除半衰期为5小时,表观体内清除率的约为1.1 mL/min/kg 。
婴儿和幼儿
单剂量给予10%口服溶液量(20 mg/kg)后,儿童患者(1个月到4岁)吸收迅速。给药1小时后血药达峰。药代动力学数据显示其半衰期(5.3小时)短于成人(7.2小时), 婴幼儿的表观体内清除率 (1.5 mL/min/kg) 快于成人(0.96 mL/min/kg) 。主要代谢产物UCB L057量,儿童低于成人。
肾功能损害患者
其左拉西坦和主要代谢产物的体内清除率取决于肌酐清除率。因此,中度或者重度肾功能不全的病人建议根据肌酐清除率调整每日维持剂量。在肾病晚期无尿症病人中,由于进行透析间期和透析期内,成人药物的血浆半衰期分别为25和3.1小时。
在4小时的透析过程中,51%左乙拉西坦被分级去除。
肝功能损害
在中轻度肝损的病人中,左乙拉西坦的清除率没有相应的变化。 大部分严重肝功损患者左乙拉西坦的清除率下降幅度大于50%,其主要原因是合并肾功能受损。
贮藏
室温(25°C或以下)贮藏
有效期
24个月
(市场价:¥297.0 每盒,500mg装,30片/盒)